化合物「GT-7」が膵臓がん細胞の細胞死を誘導することを発見 これまでの抗がん剤とは異なる作用による新たな治療法の確立に期待
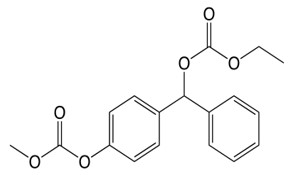
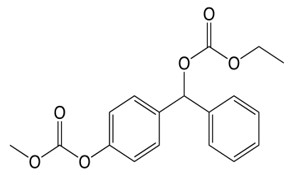
近畿大学薬学部(大阪府東大阪市)分子医療・ゲノム創薬学研究室教授 杉浦 麗子らの研究グループは、化合物「ACAGT-007a」(以下、GT-7)※1 が、特定の遺伝子変異のある膵臓がん細胞の増殖を強力に抑制し、細胞死(アポトーシス)※2 を誘導することを発見しました。さらに、細胞増殖を促進する特定の酵素の異常活性化を阻害する薬剤との併用により、その効果が増強されることも明らかにしました。本研究成果から、これまでの抗がん剤とは異なる作用を原理とした、新たな治療法の確立が期待されます。
本件に関する論文が、令和4年(2022年)2月17日(木)10:00(日本時間)、生命科学分野の国際的な学術雑誌"Cells"に掲載されました。
【本件のポイント】
●化合物GT-7が、特定の遺伝子変異のある膵臓がん細胞の細胞死を誘導することを発見
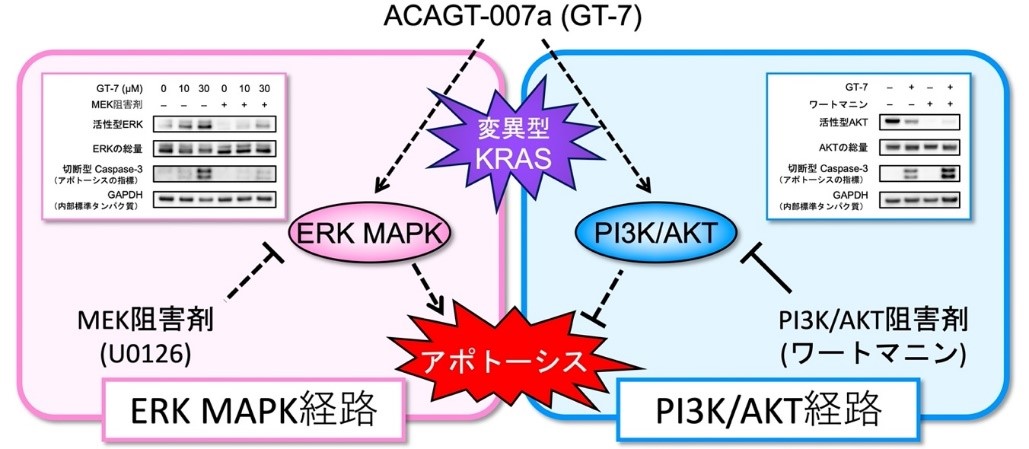
●GT-7は、「ERK MAPK」(以下、ERK)※3 という酵素の異常活性化をさらに促進することによって膵臓がん細胞の細胞死を誘導する
●GT-7と「PI3K/AKT」(以下、AKT)※4 という酵素の阻害剤との併用による膵臓がんの治療法確立に期待
【本件の背景】
細胞の増殖や細胞死には、多様なタンパク質によるシグナル伝達が関わっており、それが細胞のターンオーバーを正常に制御しています。しかし、タンパク質の遺伝子に変異が生じると細胞死が正しく誘導されなくなり、がん細胞増殖の原因となります。
90%以上の膵臓がんにおいては、KRAS※5 というタンパク質の遺伝子に変異が生じることで、細胞増殖を促進するERKとAKTという2つの酵素の働きが異常に活発になり、がん細胞の増殖を抑制できなくなると考えられています。これまでの抗がん剤開発は、ERKが関与するシグナル伝達を阻害することでがん細胞の増殖を抑制するものが中心となっていますが、膵臓がんに対する治療効果は十分でないため、別の作用を原理とした新しい治療薬の開発が強く望まれています。
【本件の内容】
近畿大学薬学部を中心とする研究グループは、特定の遺伝子変異のある膵臓がん細胞において、ERKとAKTという2つの酵素が異常に活性化していることに着目しました。今回、2つの酵素のうちERKが異常活性化しているがん細胞「メラノーマ」※6 に細胞死を誘導する化合物GT-7が、膵臓がんにも効果を示すかどうかを検証しました。
その結果、GT-7はERKの異常活性化をさらに促進することで、膵臓がん細胞の増殖を強力に抑制し、細胞死を誘導することを明らかにしました。これまでのがん治療薬はERKの活性阻害を狙うものがほとんどでしたが、GT-7はそれとは全く異なる作用によって膵臓がんに細胞死を誘導します。
また、GT-7の細胞死誘導効果は、膵臓がん細胞のAKTの働きを阻害する薬物と併用することで、顕著に増強されることも明らかになりました。本研究成果から、GT-7とAKT阻害剤の併用が、KRASタンパク質の遺伝子変異を有する特定の膵臓がんに対する新しい治療法の確立に役立つと期待されます。

【論文掲載】
掲載誌:Cells(インパクトファクター:6.6@2020)
論文名:
ACAGT-007a, an ERK MAPK Signaling Modulator, in Combination with AKT Signaling Inhibition Induces Apoptosis in KRAS Mutant Pancreatic Cancer T3M4 and MIA-Pa-Ca-2 Cells
(ERK MAPKシグナル調節薬であるACAGT-007aは、AKTシグナル阻害剤と併用することにより、KRAS変異を持つ膵臓がん細胞に対して細胞死を誘導する)
著 者:Golam Iftakhar Khandahar 1*、佐藤 亮介 1*、髙崎 輝恒 1、藤谷 佳奈 1、田邉 元三 1、坂井 和子 2、西尾 和人 2、杉浦 麗子 1
*筆頭著者
所 属:1 近畿大学薬学部、2 近畿大学医学部
【研究詳細】
研究グループは、化合物GT-7の膵臓がんへの効果を検証するにあたり、まず、KRAS遺伝子の異なる部分に変異が入った3種類の膵臓がん細胞「MIA-Pa-Ca-2」「T3M4」「PANC-1」を準備しました。それぞれの細胞にGT-7を添加したところ、GT-7の濃度依存的に細胞死が誘導され、10μMのGT-7を添加した際には、T3M4では77.1%、MIA-Pa-Ca-2では47.4%、PANC-1では32.3%の細胞に細胞死が誘導されました。また、GT-7は、すでに膵臓がん治療薬として有望視されている化合物ホノキオールより低濃度でも細胞死誘導に有効であることが分かりました。
次に、GT-7がどのように細胞死を誘導するかを検証するため、GT-7添加と同時に、ERKを活性化するタンパク質であるMEKを阻害する化合物(MEK阻害剤:U0126)を添加すると、特にT3M4においてERKの活性と細胞死が強く抑制されました。このことから、メラノーマを用いた先行研究と同様に、膵臓がん細胞でもERKをさらに活性化させることで細胞死を誘導することが明らかになりました。
また、KRASの変異によって活性化する別のシグナル伝達経路としてAKT経路があり、これを阻害する化合物としてワートマニンが知られています。ワートマニンとGT-7を同時に膵臓がん細胞に添加したところ、T3M4とMIA-Pa-Ca-2においては、誘導される細胞死の割合が顕著に増加しました。
以上の結果から、GT-7は膵臓がん細胞にあるERKをさらに活性化させることで細胞死を誘導する世界初の化合物であり、その誘導効果はAKT阻害剤と併用することによって増強されることが明らかになりました。本研究成果を活用し、特定のKRAS変異を有する膵臓がんに対して、GT-7とAKT阻害剤を併用した新たな治療法の確立が期待されます。
【用語解説】
※1 ACAGT-007a(GT-7):MAPKの活性を調節する化合物開発をめざし、ヒトと類似したシグナル伝達分子を有する分裂酵母を用いた独自の創薬探索手法により、近畿大学薬学部の研究グループが取得した化合物。メラノーマ細胞に対して、選択的に細胞死を誘導する。
※2 細胞死(アポトーシス):調節された細胞の自殺、つまりプログラムされた細胞の死滅。例えば、オタマジャクシからカエルに変態する際に尻尾がなくなるのも細胞死によるもの。臨床で用いられている抗がん剤の中には、細胞死を引き起こすことでがん細胞を死滅させる効果を持つものが多く存在する。
※3 ERK MAPK(ERK):細胞増殖を促進するシグナル伝達に関わるキナーゼ(リン酸化酵素)の一つ。膵臓がんやメラノーマなど、ある種のがんにおいてはERK MAPKが異常に活性化しており、がん化やがん細胞の増殖、転移と深く関わる。活性型RASの連鎖反応により、ERKが活性化する。
※4 PI3K/AKT(AKT):細胞増殖や代謝、タンパク質合成など様々な機能の制御に関与するセリン/スレオニンキナーゼ。細胞外からの刺激を受け、ホスファチジルイノシトール3-キナーゼ(PI3K)が活性化し、産生したPI3,4,5-三リン酸(PIP3)がAKTを活性化する。
※5 KRAS:細胞増殖を促進するシグナル伝達に関わるRASタンパク質の一つで、KRASのほかに「NRAS」、「HRAS」がある。KRASタンパク質をコードする遺伝子のいずれかに変異が起こると、異常のあるKRASタンパク質が作られ、細胞の増殖を促進するERKシグナルやAKTシグナル伝達経路が恒常的に活性化し、がんが発生しやすくなると考えられている。
※6 メラノーマ:悪性黒色腫。シミやホクロの色素成分であるメラニンを産生する細胞「メラノサイト(正常色素細胞)」が"がん化"したもの。早期に転移し、悪性度、致死率が最も高いがんの一つ。
【関連リンク】
薬学部 創薬科学科 教授 杉浦 麗子(スギウラ レイコ)
https://www.kindai.ac.jp/meikan/752-sugiura-reiko.html
薬学部 医療薬学科 教授 田邉 元三(タナベ ゲンゾウ)
https://www.kindai.ac.jp/meikan/354-tanabe-genzou.html
薬学部 創薬科学科 講師 佐藤 亮介(サトウ リョウスケ)
https://www.kindai.ac.jp/meikan/1237-satoh-ryosuke.html
薬学部 創薬科学科 講師 髙崎 輝恒(タカサキ テルアキ)
https://www.kindai.ac.jp/meikan/2084-takasaki-teruaki.html
医学部 医学科 教授 西尾 和人(ニシオ カズト)
https://www.kindai.ac.jp/meikan/757-nishio-kazuto.html
医学部 医学科 講師 坂井 和子(サカイ カズコ)
https://www.kindai.ac.jp/meikan/1674-sakai-kazuko.html
薬学部
https://www.kindai.ac.jp/pharmacy/
医学部
https://www.kindai.ac.jp/medicine/